


2-溴-3-甲基酚的应用_凯茵工业添加剂
背景及概述
2-溴-3-甲基酚为酚类衍生物,可作有机原料,用于制备医药中间体。
制备[1]

将2-氨基-间甲酚8a(5.7g,46.3mmol)溶于H2O(30mL)和1,4-二恶烷(15mL)中。将混合物加热至回流,然后在20分钟内逐滴加入HBr(48%,17mL,0.31mol)。添加完成后,将回流再保持15分钟。将反应冷却至0℃,并在30分钟内加入NaNO2的H2O(20mL)溶液。在0℃下继续搅拌15分钟,然后将混合物一次性转移至搅拌的Cu(1)Br(7.64g,53.2mmol)在H2 O(20mL)和HBr(48%,17 mL,0.31 mol)中的混合物中。在0°C(无光照)下。将反应在0℃下搅拌15分钟,温热至60℃,再搅拌15分钟,冷却至室温,然后搅拌过夜。然后将反应混合物转移到分液漏斗中并用EtOAc(3x)萃取。合并有机层,用盐水洗涤,经无水MgSO4干燥,过滤并在二氧化硅上浓缩,得到混合物,使用CombiFlash Companion(20%EtOAc /己烷)纯化,得到所需的溴化物2-溴-3-甲基酚8b(1.46g,17%产率)为红棕色油状物。
应用 [2]
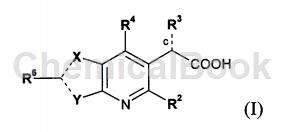
WO 2009062288提供具有抗HIV复制抑制活性的一系列新化合物。本发明的代表性化合物具有在细胞为基础的HIV复制测试中作为抑制剂的活性。本发明化合物具有对HIV整合酶的亲和力。因此,本发明化合物可用于抑制HIV整合酶的活性,且可用以降低HIV复制。本发明的其它目的对本领域技术人员可从下面说明书与实施例中得到。本发明提供式(I)化合物的异构体、外消旋体、对映异构体或非对映异构体:
硼酸酯片段8h(可偶联至噻吩并吡啶骨架,得到本发明的化合物)的合成
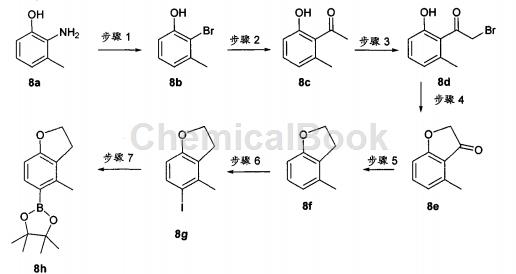
步骤1
将2-氨基-间-甲基酚8a(5.7克,46.3毫摩尔)溶于H2O(30毫升)与1,4-二噁烷(15毫升)中。将混合物加热至回流,然后经20分钟滴加HBr(48%,17毫升,0.31摩尔)。添加完成后,再回流15分钟。将反应冷却至0℃,并经30分钟添加NaNO2的H2O溶液(20毫升)。在0℃持续搅拌15分钟,接着于0℃下,将混合物以一次注射转移至Cu(I)Br(7.64克,53.2毫摩尔)在H2O(20毫升)与HBr(48%,17毫升,0.31摩尔)中的正在搅拌混合物(经保护以避光)。将反应在0℃搅拌15分钟,温热至60℃,再搅拌15分钟,冷却至室温,然后搅拌过夜。接着,将反应混合物转移至分液漏斗,且用EtOAc萃取(3x)。合并有机层,用盐水洗涤,用无水MgSO4干燥,过滤,并在硅胶上浓缩,得到混合物,其使用Companion纯化(20%EtOAc/己烷),得到所需的溴化物2-溴-3-甲基酚8b(1.46克,17%产率),为红褐色油状物。
步骤2:
于溴化物8b(1.36克,7.27毫摩尔)与(PPh3)2PdCl2(766毫克,1.09毫摩尔)在DMF(12毫升)中的溶液内,添加1-乙氧基乙烯基-三-正-丁基锡(2.7毫升,8.0毫摩尔)。将混合物加盖,并在微波中,于160℃加热15分钟。HPLC和LC-MS分析显示大约70%转化率。添加更多的1-乙氧基乙烯基-三-正-丁基锡(2.7毫升;8.0毫摩尔)与催化剂(PPh3)2PdCl2(380毫克,0.05摩尔%),并将溶液再次接受相同微波条件。用6N HCl(1.5毫升)将反应终止,并于室温搅拌1小时,至达成中间体的水解作用。将混合物倒入EtOAc(150毫升)中,用盐水洗涤(3x),用MgSO4干燥,过滤,并在硅胶上浓缩,得到混合物,其使用Companion纯化,得到所需的酮8c(947毫克,87%产率),为橙色油状物。
步骤3:
将甲基酮8c(1.02克,6.8毫摩尔)溶于EtOAc(15毫升)与CHCl3(15毫升)中,然后用Cu(II)Br2(3.03克,13.6毫摩尔)处理。将混合物加热至回流,持续16小时。将混合物冷却至室温,过滤产物,并用EtOAc洗涤(1x)。将溶液于硅胶上浓缩,得到混合物,其使用Companion纯化(10%EtOAc/己烷),得到α-溴代酮8d(710毫克,46%产率),为橙色油状物。将此物质以本身使用于下一步骤,无需纯化。
步骤4:
于溴代酮8d(710毫克,3.1毫摩尔)在无水DMF(12毫升)中的溶液内,添加KF(400毫克,6.95毫摩尔)。将反应在室温搅拌16小时。将混合物溶于EtOAc(150毫升)中,用盐水洗涤(3x),用无水MgSO4干燥,过滤,并在硅胶上浓缩,得到混合物,其使用Companion纯化(20%EtOAc/己烷),得到环酮8e(280毫克,61%产率),为淡橙色固体。
步骤5:
Zn粉预活化方法:将锌粉(20克,350目)置于圆底烧瓶中,并添加1N HCl(50毫升)。将该悬浮液超声1分钟,然后倾析出液体。重复此步骤两次,接着,将固体用EtOH(2x)、Et2O(2x)洗涤,并在高真空下干燥。于酮8e(280毫克,1.89毫摩尔)在AcOH(10毫升)中的溶液内,添加预活化的Zn粉(1.24克,18.9毫摩尔)。然后,将反应混合物加热至75℃,持续2小时。过滤反应混合物(用EtOAc洗涤固体)。于硅胶上蒸发溶剂,且混合物使用Companion直接纯化(10%EtOAc/己烷),得到所需的二氢并呋喃8f(174毫克,69%产率),为无色油状物。
步骤6:
于二氢并呋喃8f(240毫克,1.8毫摩尔)在MeOH(5毫升)中的溶液内,添加AgNO3(304毫克,1.79毫摩尔),接着添加碘(453毫克,1.79毫摩尔)。将黄色混合物在室温搅拌1小时。于反应混合物中,添加10%Na2S2O3溶液,并将混合物在室温搅拌15分钟。用EtOAc(100毫升)稀释混合物,并将有机层用盐水(3x)与10%Na2S2O3(2x)洗涤。将有机相用无水MgSO4干燥,过滤,并在硅胶上浓缩,得到混合物。将此混合物使用Companion纯化(10%EtOAc/己烷),得到碘代衍生物8g(400毫克,86%产率),为白色无定形固体。
步骤7:
将碘代衍生物8g(400毫克,1.54毫摩尔)、双(频哪醇)二硼烷(585毫克,2.31毫摩尔)、钾(511毫克,5.4毫摩尔)在DMF(20毫升)中的混合物脱氧(Ar气球,并超声5分钟);然后添加催化剂(PdCl2dppf,188毫克,0.23毫摩尔),伴随着再脱气(Ar气球,并超声2分钟)。接着,将混合物加热至大约95℃,持续4小时。将混合物冷却,添加EtOAc(200毫升),用盐水(3x)、水(2x)洗涤,用无水MgSO4干燥,过滤,并在硅胶上溶剂蒸发,得到混合物,其使用Companion纯化(10%EtOAc/己烷),得到所需的硼酸酯8h(315毫克,79%产率),为黄色油状物。
主要参考资料
[1] PCT Int. Appl., 2009062285, 22 May 2009
[2] PCT Int. Appl., 2009062288, 22 May 2009